Множинна мієлома. Діагностика та лікування
Множинна мієлома (ММ) – це злоякісне лімфопроліферативне захворювання з В-лімфоцитів (плазматичних клітин), що характеризується проліферацією та акумуляцією плазматичних клітин, головним чином, у кістковому мозку та надмірною продукцією моноклональних імуноглобулінів, які виявляють у плазмі крові і сечі, деструктивним ураженням скелета, розвитком ниркової недостатності, гіперкальціємії та анемії.
 Множинна мієлома є пухлиною групи імуноглобулін-секретуючих лімфом. Вона зустрічається з частотою, що становить 1% від усіх онкологічних і 14% – онкогематологічних захворювань. Середня захворюваність на ММ у світі становить 4 випадки на 100 тисяч населення. Найвищу захворюваність на ММ спостерігають серед чоловіків негроїдної раси в США (1 випадок на 10 тис мешканців), а найнижчу – в Китаї (1 випадок на 100 тисяч населення). Останнім часом у світі відмічають тенденцію до зростання захворюваності на ММ.
Множинна мієлома є пухлиною групи імуноглобулін-секретуючих лімфом. Вона зустрічається з частотою, що становить 1% від усіх онкологічних і 14% – онкогематологічних захворювань. Середня захворюваність на ММ у світі становить 4 випадки на 100 тисяч населення. Найвищу захворюваність на ММ спостерігають серед чоловіків негроїдної раси в США (1 випадок на 10 тис мешканців), а найнижчу – в Китаї (1 випадок на 100 тисяч населення). Останнім часом у світі відмічають тенденцію до зростання захворюваності на ММ.
В Європі частота виявлення множинних мієлом становить 6 випадків на 100 тис населення щорічно. Переважно на множинні мієломи хворіють люди старшого віку (максимум захворюваності припадає на вік 50–70 років). Водночас реєструють захворювання, починаючи з 18-річного віку. Пацієнти до 40 років становлять 5–10%. Чоловіки хворіють частіше, ніж жінки (співвідношення 3:2).

В Україні впродовж останніх років захворюваність на множинну мієлому становить 2,4 випадка на 100 тисяч населення. Згідно з даними МОЗ України, в 2008 році зареєстровано понад 2500 пацієнтів із множинною мієломою. Проблема повного виліковування хворих на ММ є нерозв’язаною. До недавнього часу повних тривалих ремісій досягали вкрай рідко. Без лікування середня тривалість життя становила 6 місяців. У разі проведення стандартної хіміотерапії МР (мелфалан – преднізолон) даний показник збільшується до 3 років. За даними національного канцеррегістру в Україні у 2008 році 37,9% хворих на ММ померли впродовж першого року з моменту встановлення діагнозу. Нині в Європі лише близько 35% пацієнтів із ММ дожили до п’яти років від дати встановлення діагнозу. Середньоєвропейський показник смертності від даного захворювання становить 4 на 100 тисяч населення на рік.
Проводити лікування хворого на множинну мієлому повинні лікарі-гематологи із залученням спеціалістів інших спеціальностей. Кваліфікована допомога такому пацієнту полягає в наданні спеціалізованих видів діагностики та лікування, вона повинна здійснюватися за чіткою схемою.
Так, під час діагностично-лікувального процесу може виникнути потреба в розгорнутому гематологічному дослідженні, в тому числі патоморфологічне, у проведенні специфічних біохімічних тестів, променевої діагностики, а також у наданні деяких видів спеціалізованої допомоги – нефрологічної (в т.ч. можливість швидкого проведення діалізу), онкологічної (в т.ч. променева терапія), хірургічної, ортопедичної, нейрохірургічної. Зрештою, під час визначення показань до трансплантації кісткового мозку та стовбурових клітин крові необхідною умовою є наявність акредитованого центру трансплантації.
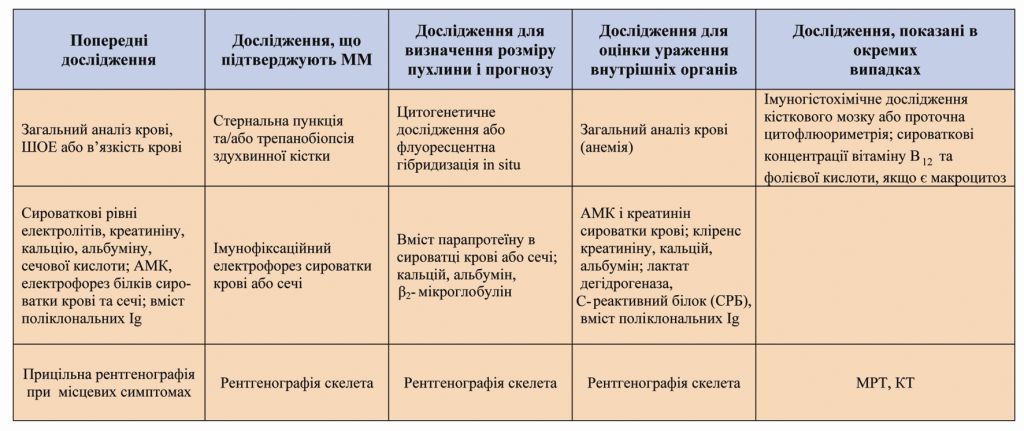
Зазвичай, множинну мієлому діагностують у разі виявлення парапротеїну в сироватці крові або сечі, а також за наявності остеолітичних вогнищ під час проведення рентгенологічного дослідження в сукупності із збільшенням кількості плазматичних клітин у кістковому мозку. Перелік основних клінічних, лабораторних та інструментальних тестів, необхідних для встановлення діагнозу ММ та його верифікації, наводимо в табл. 1.
Таблиця 1. Види обстеження хворих на ММ
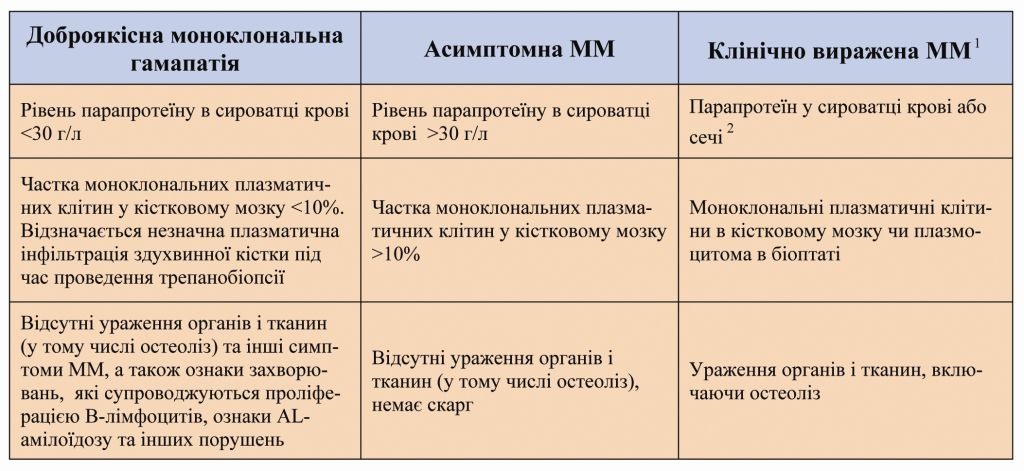
 Під час проведення діагностики множинних мієлом важливо диференціювати її від інших захворювань, які також супроводжуються появою парапротеїну в крові та сечі. Серед таких патологій – доброякісна моноклональна гамапатія, AL-амілоїдоз, солітарна плазмоцитома (кісткова або в м’яких тканинах), В-клітинні лімфоми (у тому числі макроглобулінемія Вальденстрема), хронічний лімфолейкоз. Незначна кількість парапротеїну в сечі та зниження вмісту поліклональних імуноглобулінів не є обов’язковими симптомами ММ, вони можуть свідчити про розвиток доброякісної моноклональної гамапатії (Kyle et al., 2002). Ця патологія досить поширена: за статистичними даними, її реєструють у 2% осіб віком понад 55 років і в 3% – понад 70 років. Тобто вміст невеликої кількості парапротеїну в сироватці крові з більшою вірогідністю вказує на доброякісну гамапатію, а не ММ. Для чіткого розмежування цих патологій International Myeloma Working Group в 2003 р. створила систему критеріїв диференційного діагнозу (табл. 2).
Під час проведення діагностики множинних мієлом важливо диференціювати її від інших захворювань, які також супроводжуються появою парапротеїну в крові та сечі. Серед таких патологій – доброякісна моноклональна гамапатія, AL-амілоїдоз, солітарна плазмоцитома (кісткова або в м’яких тканинах), В-клітинні лімфоми (у тому числі макроглобулінемія Вальденстрема), хронічний лімфолейкоз. Незначна кількість парапротеїну в сечі та зниження вмісту поліклональних імуноглобулінів не є обов’язковими симптомами ММ, вони можуть свідчити про розвиток доброякісної моноклональної гамапатії (Kyle et al., 2002). Ця патологія досить поширена: за статистичними даними, її реєструють у 2% осіб віком понад 55 років і в 3% – понад 70 років. Тобто вміст невеликої кількості парапротеїну в сироватці крові з більшою вірогідністю вказує на доброякісну гамапатію, а не ММ. Для чіткого розмежування цих патологій International Myeloma Working Group в 2003 р. створила систему критеріїв диференційного діагнозу (табл. 2).
Таблиця 2. Діагностичні критерії доброякісної моноклональної гамапатії та ММ (International Myeloma Working Group, 2003)
 ¹ За наявності доброякісної моноклональної гамапатії можуть спостерігати AL-амілоїдоз та Ig M-парапротеїнемічну нейропатію.
¹ За наявності доброякісної моноклональної гамапатії можуть спостерігати AL-амілоїдоз та Ig M-парапротеїнемічну нейропатію.
² Іноді рівень парапротеїну не має діагностичного значення. У деяких випадках його не виявляють ні в сироватці крові, ні в сечі, проте наявні ураження органів і тканин, збільшений вміст плазматичних клітин у кістковому мозку (несекретуюча форма ММ).
³ При ураженні кісток і тканин навіть за відсутності скарг діагностують клінічно виражену ММ. Такі хворі потребують термінового початку терапії.
Хіміотерапія та інші види лікування показані за наявності клінічно вираженої множинної мієломи, а також асимптомної форми з ураженням органів і тканин. Дані низки рандомізованих контрольованих досліджень свідчать про те, що в інших випадках за наявності асимптомної ММ раннє проведення хіміотерапії неефективне (Hjorth et al., 1993; Riccardi et al., 2000). Єдиним достовірним прогностичним чинником переродження доброякісної гамапатії на ММ чи В-клітинну лімфому є рівень парапротеїну в сироватці крові (Kyle et al., 2002; Kyle, Rajkumar, 2003).
Вірогідність виникнення бактеріємії за наявності гіпогамаглобулінемії у хворих із доброякісною гамапатією вдвічі більша, ніж у здорової людини. Спостереження за пацієнтами з доброякісною гамапатією та асимптомною формою ММ повинно бути тривалим. Частота проведення періодичних оглядів залежить від ризику прогресування. Цей ризик визначається рівнем парапротеїну (рівень III, клас В).
За наявності асимптомної множинної мієломи при клінічному спостереженні повинні проводитися регулярні огляди (1 раз на 3 міс) із визначенням рівня парапротеїну в сироватці крові та сечі. Дослідження кісткового мозку та рентгенографію скелета здійснюють за умови появи нових симптомів захворювання (рівень IV, клас С).
За наявності доброякісної моноклональної гамапатії під час спостереження потрібно також здійснювати регулярні огляди з визначенням рівня парапротеїну в сироватці крові; за умови низького ризику прогресування інтервали між оглядами мають становити 6–12 міс (рівень IV, клас С).
Лікування починають лише за умови прогресування захворювання або за наявності ознак ураження органів і тканин (рівень Ib, клас А).
Рентгенологічні ознаки ураження кісток, навіть за відсутності інших клінічних симптомів, є показанням до термінового початку лікування (рівень IIb, клас В). Нині в таких випадках встановлюють діагноз клінічно вираженої множинної мієломи.
Високі рівні b2 -мікроглобуліну, альбуміну, СРБ в сироватці крові, а також гіпоальбумінемія належать до несприятливих прогностичних ознак. Атипові плазматичні клітини та високий проліферативний індекс також вказують на несприятливий прогноз. Серед важливих чинників, які визначають перебіг захворювання, варто назвати вид хромосомної аномалії. Прогноз істотно погіршується в разі моносомії за 13-ю хромосомою або делеції 13-ї хромосоми, гіпо-, псевдодиплоїдному або тетраплоїдному каріотипі та деяких компенсованих транслокаціях t (4; 14), t (14; 16) (Fonseca et al., 2004).
З часу появи класифікації Durie, Salmon (1975) було здійснено багато спроб її вдосконалити. Зокрема, до існуючих критеріїв додавали визначення рівня b2-мікроглобуліну та концентрації альбуміну в сироватці крові, які ця класифікація не враховує. У 2003 р. робоча група, очолювана Greipp et al., запропонувала міжнародний прогностичний індекс, який на підставі визначення цих показників дає можливість робити прогноз для кожного хворого з ММ, незалежно від терапії (табл. 3). Цитогенетичні дані підвищують точність визначення стадії ММ за цією системою.
Таблиця 3 Міжнародна система стадіювання (ISS) ММ
 Серед завдань з лікування за однією зі схем виділяють:
Серед завдань з лікування за однією зі схем виділяють:
– пригнічення росту пухлини;
– максимальне поліпшення якості життя;
– збільшення тривалості життя.
Виконати ці завдання можна шляхом проведення хіміо- та підтримувальної терапії. Тактика лікування визначається індивідуально. За можливістю призначають терапію в режимі МР або високодозову хіміотерапію з подальшою алогенною трансплантацією кісткового мозку чи стовбурових клітин крові. Однак у більшості хворих не може бути застосоване таке лікування через похилий вік, супутні захворювання або тяжкий загальний стан. Важливо, щоб до початку лікування був розроблений його детальний план із урахуванням виникнення рецидиву.
Чутливість до стандартних схем хіміотерапії є одним із важливих прогностичних критеріїв при ММ. Нові можливості подолання генетично обумовленої резистентності до хіміотерапії з’явилися з появою нових препаратів. Їх висока ефективність доведена за допомогою рандомізованих клінічних досліджень у разі лікування пацієнтів з різними стадіями ММ, насамперед, ІІ та ІІІ стадіями – SUMMIT, APEX, VISTA та інші. Результати означених досліджень доводять, що бортезоміб необхідно розглядати як основний компонент нових стандартних схем терапії для пацієнтів із ММ, яким не планується трансплантація кісткового мозку. Широкому впровадженню препарата бортезоміб у практику лікування хворих на ММ перешкоджає його висока вартість. У більшості держав існує практика гарантованого фінансового забезпечення життєво необхідними онкологічними препаратами за умови доведеної ефективності та економічної доцільності. Однак слід пам’ятати, що онкологічні препарати належать до життєво необхідних, тому повинні бути доступними не тільки пацієнтам, що можуть сплатити їх вартість. В Україні було вивчено клініко-економічну доцільність застосування бортезомібу при ММ. Автором було проведено порівняльний фармакоекономічний аналіз різноманітних схем хіміотерапії ММ методом «затрати/ефективність» із застосуванням моделювання. Аналізувалися прямі медичні затрати на медикаментозну терапію у разі застосування наступних схем хіміотерапії: стандартна схема МР (мелфалан – преднізолон), (бортезоміб – мелфалан – преднізолон), VMP (бортезоміб – талідомід – преднізолон), VMPT (бортезоміб – мелфалан – преднізолон – талідомід) та ТМР (талідомід – мелфалан – преднізолон). В якості кінцевого критерію ефективності схеми лікування розглядалися тривалість життя хворих на ММ. Відмічено, що в разі застосування всіх схем хіміотерапії тривалість життя хворих обмежувалася 2-3 роками. Автор підкреслила, що в якості сурогатного критерію тривалості життя під час проведення рандомізованих клінічних досліджень може використовуватися медіана виживання, а також частина хворих, які вижили впродовж певного часу одного, двох або трьох років, проте, на жаль, вони залежать від часу спостереження за хворими, а тому можуть істотно відрізнятися.
На підставі ретроспективного аналізу результатів лікування 759 пацієнтів з ММ в період 1987–2007 рр. М. Wang і співавт. (2008) зробили висновок, що збільшення тривалості життя означених хворих тісно пов’язане з рівнем відповіді на проведене лікування. Досягнення повної відповіді дозволяє істотно продовжити життя хворим – медіана виживання цієї групи хворих 9,6 року, водночас, за неповної відповіді і відсутності відповіді на терапію становили відповідно 4,0 і 2,2 року. Медіана виживання у хворих на ММ з повною відповіддю не залежала від того, за якою схемою лікування було одержано відповідь.
Як свідчать результати досліджень Н.В. Бездетко (2010), найбільш дешева схема хіміотерапії – МР – характеризується дуже низькою ефективністю порівняно з усіма альтернативними. Ефективність схеми МРТ також істотно, в 1,8 разу нижча, ніж схеми VMP i VTP. Відмінності в ефективності між схемами VMP i VTP є незначними, як і відмінності у їх вартості. Найбільший приріст тривалості життя дозволяє одержати схема VMPT, проте вона водночас є найбільш вартісною – майже вдвічі дорожче, ніж схеми VMP i VTP. Один додатковий рік життя у разі переходу зі схеми МЗ на схему VMP потребує збільшення витрат на кожного хворого на 2746 грн, на схему VTP – 2561 грн.
Препарати нового покоління надають можливість значно продовжити тривалість життя після першого рецидиву. Результати дослідження VISTA дозволяють зробити припущення, що незабаром будуть внесені зміни у схему застосування бортезомібу і аналогів талідоміду (леналідомід).
Автогенна трансплантація стовбурових клітин крові (ASCT). АSСТ поліпшує показники досягнення повної відповіді та продовжує медіану загальної виживаності за наявності ММ приблизно на 12 міс. Показник летальності становить 1-2%.
Послідовна трансплантація. Під час послідовної (подвійної) ASCT пацієнти отримують другу заплановану ASCT після відновлення від першої процедури. Нещодавно було отримано результати рандомізованого дослідження Intergroupe Francophone du Myelome (IFM), які вказують на значне підвищення загальної виживаності та безрецидивної виживаності у групах пацієнтів, які отримали подвійну ASCT. Подібні результати були продемонстровані у рандомізованому дослідженні, проведеному в Італії. Нині опубліковані результати двох рандомізованих досліджень, які не показали значного поліпшення показників загальної виживаності за послідовної ASCT, однак ці випробування мали порівняно короткий період наступного спостереження.
В обох дослідженнях – французькому та італійському – доведено переваги проведення другої ASCT для пацієнтів, у яких не було досягнуто повної або стійкої часткової відповіді (зниження рівня М-білка на 90% і більше ) після першої процедури. На підставі цих даних продовжується збір достатньої кількості стовбурових клітин для хворих, які відповідають встановленим критеріям для проходження трансплантації двічі. Пацієнтам, у яких вдалося отримати хорошу відповідь після першої трансплантації, рекомендовано моніторинг; також пропонується участь у клінічних дослідженнях із вивчення підтримувальної терапії. Водночас для них резервується матеріал для другої ASCT на випадок рецидиву. Пацієнтам, які не продемонстрували повної або доброї часткової відповіді після першої трансплантації, пропонується друга ASCT.
Алогенна трансплантація. Перевагами алогенної трансплантації є відсутність контамінації трансплантата пухлинними клітинами та наявність ефекту «трансплантат проти пухлини». Однак лише 5–10% пацієнтів є кандидатами за такими критеріями: вік, наявність НLA-сумісного донора, адекватний функціональний стан органів і систем. Крім того, висока частота летальних випадків, пов’язаних із лікуванням, що зумовлені, насамперед, реакцією «трансплантат проти хазяїна», робить алогенну трансплантацію неприйнятною для більшості пацієнтів із ММ.
Тривають дослідження ефективності використання міні-алогенної трансплантації. Найкращі результати міні-алогенної трансплантації з використанням матеріалу НLA-ідентичного донора були відзначені у пацієнтів з недавно діагностованою ММ. Летальність, пов’язана з проведенням маніпуляції, становить 10–15%.
Підтримувальна терапія інтерфероном альфа-2 має обмежене застосування та використовується рідко. Дані дослідження Berenson et al. свідчать, що під час її проведення застосування преднізолону дозволяє збільшити показники безрецидивної (14 проти 5 міс) і загальної виживаності (37 проти 26 міс). Однак такі результати було отримано у пацієнтів, яким не проводили АSСТ і в яких від початку була отримана добра відповідь на терапію глюкокортикостероїдами. Тому узагальнення цих результатів на практиці є складним завданням.
Нині проводять клінічні дослідження оцінки ефективності талідоміду, вакцинації дендритними клітинами та інших нових методів підтримувальної терапії ММ. Поліпшення показників безрецидивної виживаності на тлі терапії талідомідом і памідронатом було отримано французькими дослідниками (IFМ 99-02), але збільшення загальної виживаності дотепер не спостерігали.
Для пацієнтів з ММ високого ризику характерна резистентність до більшості наведених схем лікування. У них може відзначатися відсутність відповіді на лікування навіть у разі використання послідовної АSСТ. У лікуванні таких хворих доцільно розглядати можливість застосування новітніх терапевтичних стратегій. Більшість рекомендацій щодо ведення пацієнтів із резистентною ММ базується винятково на думках експертів (рівень IV, клас С). Традиційно нові засоби розглядають як основу первинної терапії на ранніх стадіях захворювання, а АSСТ залишають на випадок рецидиву. Як альтернативу пропонують проведення АSСТ з послідовним застосуванням алогенної трансплантації. Однак дослідження IFМ, в тому числі пацієнти з делецією 13-ї хромосоми та високим рівнем мікроглобуліну, не продемонструвало переваг цієї стратегії порівняно з послідовною АSСТ.
Лікування рецидиву. Якщо рецидив відбувається через 6 міс після закінчення терапії, то необхідно призначити повторний курс хіміотерапії в аналогічному режимі. Пацієнтам, для яких на ранньому етапі захворювання були зібрані стовбурові клітини і збережені шляхом кріоконсервування, можна проводити АSСТ.
Якщо рецидив характеризується кволим перебігом, доцільно проводити лікування в режимі МР або навіть у монорежимі з використанням цих агентів. У таких випадках, зазвичай, спостерігають асимптомне збільшення рівня М-білка в сироватці крові та сечі, помірну анемію або незначні літичні ураження кісток. Навпаки, пацієнтам із агресивним рецидивом необхідно одразу призначати комбіновану терапію. Хворі з рецидивуючою ММ, зазвичай, продовжують приймати один лікарський препарат або їх комбінацію до появи рецидиву або проявів токсичності. В останньому випадку їм призначають такий варіант терапії: високі дози дексаметазону або метилпреднізолону є прийнятною альтернативою в лікуванні рецидивуючої ММ, особливо якщо пацієнт добре відповідав на початкову терапію глюкокортикостероїдами та не застосовував високі дози даних препаратів на момент рецидиву.
Пацієнтів, які не є кандидатами для трансплантації на підставі віку, статусу прояву і супутнього захворювання, лікують стандартною терапією алкілювальними сполуками. Найчастіше надають перевагу пероральному МР. Цей режим характеризується мінімальною токсичністю. Незважаючи на кращі показники досягнення повної відповіді, не було продемонстровано переваг у показниках виживаності під час застосування кожного з більш агресивних комбінованих режимів хіміотерапії порівняно з МР (мелфалан + преднізолон). Мелфалан використовують у дозі від 8 до 10 мг/день із преднізолоном 60 мг/день перорально протягом 7 днів з повторним курсом кожні 6 тижнів. Загальний аналіз крові контролюють кожні 3 тижні після початку терапії. Відповідно до отриманих показників дозу мелфалану коригують таким чином, щоб цитопенія в середині циклу була помірною. Застосування лише дексаметазону, Тhal/Dex або інших індукційних режимів, доцільних у кандидатів на трансплантацію, може розглядатися як початкова терапія для пацієнтів, які не є кандидатами на АSСТ. Однак ці режими мають більшу токсичність, не демонструють переваг у показниках виживаності порівняно з МР. Використання цих схем, а також режимів комбінованої хіміотерапії доцільне тоді, коли бажано безпосередньо скоротити пухлинну масу за наявності великих літичних уражень кісткової тканини або ниркової недостатності. За умови контрольованого перебігу захворювання можливий перехід із комбінованого режиму терапії на МР. Режим із застосуванням бортезомібу – оптимальна альтернатива для пацієнтів, які не є кандидатами на АSСТ.
Нині міжнародна медична спільнота вважає економічно доцільними ті медичні втручання, за яких ціна додаткового місяця життя становить від 1000 до 5000 євро. Перехід від класичної схеми МР на значно ефективніші схеми терапії потребують для отримання додаткового року життя близько 200 євро на одного хворого. Це істотно менше, ніж допускають міжнародні стандарти, і є доцільним, навіть в умовах обмеженого фінансування.
Станіслав ВИДИБОРЕЦЬ
професор кафедри гематології та трансфузіології НМАПО імені П.Л.Шупика
Ніна КОСТЮКОВА
аспірант кафедри гематології та трансфузіології НМАПО імені П.Л. Шупика
Київський міський центр трансплантації кісткового мозку
Бажаєте знати більше – прочитайте:
1. Авксентьєва М.В., Воробьев П.А. Клинико-экономический анализ применения бортезомиба (велкейд) при множественной миеломе // Проблемы стандартизации в здравоохранении. – 2007. – № 4.– С. 7-10.
2. Бездетко Н.В. Фармакоэкономический анализ применения препарата велкейд (бортезомиб) в лечении множественной миеломы // Здоров’я України – 2010, №1 (8). – С. 22-23.
3. Бессмельцев С.С., Абдулкадыров К.М. Множественная миелома. С-Пб.: Диалект, 2004. – 446 с.
4. Войцеховский В.В. Анализ результатов лечения больных множественной миеломой /В.В. Войцеховский, Ю.С. Ландышев, В.В. Есенин и др. // Дальневосточный медицинский журнал. – 2007. – № 1. – С. 47-50.
5. Войцеховский В.В. Лечение пациентов с резистентной и рецидивирующей формами множественной миеломы препаратом велкейд (бортезомиб) / В.В. Войцеховский, Ю.С. Ландышев, В.В. Есенин и др. // Нове в гематології та трансфузіології. – К., 2007. – Вип. 7. – С. 88-94.
6. Вотякова О.М., Демина Е.А. Множественная миелома. В кн.: Клиническая онкогематология / Под ред. М.А. Волковой. – 2-е изд., перераб. и доп. – М.: ОАО «Издательство «Медицина», 2007. – С. 847-873.
7. Вотякова О.М. Множественная миелома: достижения лекарственного лечения ХХ-ХХІ веков // Онкогематология. – 2004. – Т. 6, № 4. – С. 19-25.
8. Вотякова О.М. Современная терапия множественной миеломы // Бюллетень сибирской медицины. – 2008. – № 3. – С. 33-41.
9. Всеукраїнська громадська організація «Асоціація допомоги інвалідам та пацієнтам з ХЛПЗ (хронічні лімфопроліферативні захворювання)».
10. Змачинский В.А. Лечение множественной миеломы. Опыт Белорусского центра гематологии и трансплантации костного мозга / В.А. Змачинский, А.Л. Усс, Н.Ф. Миланович и др // Гематология и трансфузиология. – 2005. – № 6 – С. 45-48.
11. Ландышев Ю.С., Войцеховский В.В. Современные аспекты диагностики и лечения множественной миеломы / Санк-Петербургские врачебные новости. – 2006. – № 4. – С. 18-22.
12. Матлан В.Л. Миеломная болезнь // Мистецтво лікування. – 2006. – № 1. – С. 27-30.
13. Моисеев С.И., Салогуб Г.Н., Степанова Н.В. Современные принципы диагностики и лечения множественной миеломы. – СПб: Издательство СПбГМУ, 2006. – 39 с.