Гемофілія. Гемартроз – найчастіший прояв гемофілії
Гемофілія як найчастіша і найтяжча форма спадкових коагулопатій не тільки належить до складних медичних проблем, а й має важливий соціальний аспект. Ця хвороба призводить до ранньої втрати працездатності та інвалідизації більшості хворих, переважно ще у дитячому віці.
Хворі на гемофілію потребують значних матеріальних затрат на постійну замісну трансфузійну терапію препаратами плазми крові при повторних кровотечах, а також створення особливих умов навчання і роботи. Ураження опорно-рухового апарату є показами до оперативного лікування та повноцінної реабілітації, а в разі інвалідизації хворі потребують стороннього догляду.
Як свідчить практика, організація з надання медичної допомоги в Україні не відповідає сучасним світовим стандартам. Доцільно: впровадити профілактичне лікування у хворих на гемофілію (виділивши пріоритетні групи – діти з важкими формами гемофілії), проводити лікування хворих «на дому», створити регіональні центри гемофілії та інших коагулопатій і окреслити їх завдання, структуру, обов’язки та права, передбачити відповідне фінансування та матеріальне забезпечення. Окремо стоїть питання професійно-медичної реабілітації, а також санаторно-курортного лікування інвалідів, хворих на гемофілію.
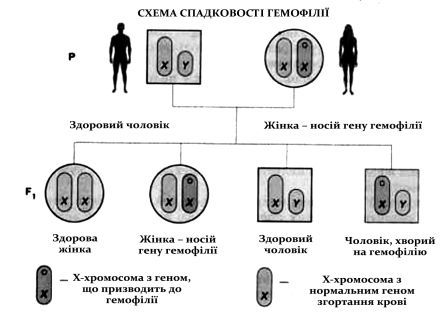
Розповсюдженість гемофілії А становить 1:10000, гемофілії В – 1:30000-50000 жителів чоловічої статі. Гемофілія успадковується за рецесивною ознакою, зчепленою зі статевою Х-хромосомою, при цьому успадковуються один і той самий тип гемофілії та однакова тяжкість захворювання. У загальній популяції хворих на гемофілію 30-40% випадків припадають на спорадичну гемофілію, зумовлену патологічною мутацією гена.
В Україні станом на початок 2008 р. на диспансерному обліку перебувало 2456 хворих на гемофілії та хворобу Віллебранда, з них 664 (27%) дітей. Серед них: гемофілія А (дефіцит VIIІ фактора), діагностована у 1264 дорослих і 464 дітей, а гемофілія В (дефіцит ІХ фактора) – у 190 дорослих і 81 дітей.
Близько 70% хворих страждають від тяжких і середньотяжких форм перебігу гемофілії, за яких ураження опорно-рухового апарату мають прогресуючий характер і є основною причиною ранньої інвалідизації.
Наразі картовано гени, що контролюють синтез факторів VIII або IX і відповідні за розвиток гемофілії. Ген, що кодує синтез фактора VIII (186 тисяч пар нуклеотидів), розташовано на довгому плечі Х-хромосоми у локусі Xq 28, він складається з 26 екзонів і 25 інтронів і містить 2332 амінокислоти. Ген фактора ІХ (34 тисячі пар нуклеотидів) розташовано у локусі Xq 27 довгого плеча Х-хромосоми та складається з 8 екзонів.
Класифікація гемофілії (МКБ-10):
– Код D66. Спадковий дефіцит фактора VIII (Гемофілія А).
– Код D67. Спадковий дефіцит фактора ІХ (Гемофілія В).
Активність факторів VIII і ІХ варіює у загальній популяції від 50 до 200%. Тяжкість клінічного перебігу захворювання залежить від рівня фактора VIII або ІХ, що дозволяє класифікувати захворювання таким чином:
• Тяжка форма – рівень фактора до 1%. Геморагічний синдром проявляється у ранньому дитячому віці та характеризується частими післятравматичними та спонтанними крововиливами до суглобів, м’язів, внутрішніх органів. У деяких хворих одразу після народження відзначають кефалогематому, мелену, тривалу кровотечу з пуповини. У подальшому – тривалі відстрочені кровотечі у ході прорізування та зміні молочних зубів.
• Середньотяжка форма – рівень фактора від 1 до 5%. Характеризується проявом хвороби у дошкільному віці (4-6 років) і пізніше, з помірно вираженим геморагічним синдромом, із крововиливами до суглобів, м’язів, гематурією. Усі наведені симптоми спостерігаються значно рідше, ніж за тяжкої форми. Загострення геморагічного діатезу від-значаються від 2 до 3 разів на рік.
• Легка форма – рівень фактора понад 5%. Відзначається тим, що кровотечі бувають рідше і вони менш інтенсивні. Клінічна симптоматика проявляється у шкільному віці, після травми або операції.
Поява інгібіторів фактора VIII або ІХ зсідання – одне з найтяжчих ускладнень замісної терапії у хворих на гемофілію. Під дією інгібітора екзогенний фактор VIII швидко втрачає прокоагулянтну активність, стимулює додаткову продукцію антитіл – підвищується титр і активність інгібітора у циркулюючій крові. Кровотеча набуває неконтрольованого характеру.
Антитіла до фактора VIII трапляться значно частіше (10-35%), ніж до чинника ІХ (3-5%). Вони виникають в основному в дитячому віці (3-6 років). Надалі ймовірність виникнення інгібіторів значно зменшується.
 Утворення інгібіторів даних чинників зсідання, можливо, зумовлено існуванням генетичного поліморфізму нормального антигемофільного чинника та наявністю у хворого імунореактивної, біологічно дефектної молекули. Наявність інгібітора викликає певну інверсію у перебігу захворювання, поглиблюючи його тяжкість. Важлива роль у цьому належить малій ефективності гемостатичної терапії.
Утворення інгібіторів даних чинників зсідання, можливо, зумовлено існуванням генетичного поліморфізму нормального антигемофільного чинника та наявністю у хворого імунореактивної, біологічно дефектної молекули. Наявність інгібітора викликає певну інверсію у перебігу захворювання, поглиблюючи його тяжкість. Важлива роль у цьому належить малій ефективності гемостатичної терапії.
Інгібітори чинників зсідання крові виникають не лише у хворих на гемофілію. Вони можуть утворюватись у людей, які раніше не мали геморагічного діатезу. Частіше їх виявляють в осіб зрілого віку, включаючи вагітних, і у людей похилого віку без очевидних захворювань, або у пацієнтів із різними імунними захворюваннями, такими як системний червоний вовчак, ревматоїдний артрит, регіональний ентерит тощо. Етіологія їх розвитку залишається незрозумілою.
Природа та ступінь кровоточивості відрізняються у пацієнтів із набутою та вродженою формами гемофілії. У хворих із вродженим геморагічним діатезом відзначають переважно крововиливи до суглобів, м’язів та інших м’яких тканин. Навпаки, у хворих зі спонтанно виниклим інгібітором гемартрози відзначають рідко, характерною є поява спонтанних гематом, тяжкої гематурії, ретрофарингеальних і ретроперитонеальних гематом, крововиливів до мозку.
Принципи лікування хворих із вродженою гемофілією, ускладненою інгібітором, та автоімунною гемофілією не відрізняються один від одного, але принципово відрізняються від таких для класичної гемофілії без інгібітора.
Найхарактернішим і найспецифічнішим симптомом гемофілії є крововиливи до крупних суглобів – гемартрози. Відзначаються часті внутрішньом’язові та заочеревинні гематоми, тривалі кровотечі у разі травм, видалення зубів, операцій. Рідше трапляються крововиливи до органів черевної порожнини, шлунково-кишкові кровотечі, гематурія, внутрішньочерепні геморагії.
У перші роки життя часто трапляються кровотечі зі слизових оболонок ротової порожнини, носові кровотечі, синці на шкірі. Тяжчий перебіг діатезу відзначається у періоди або невдовзі після перенесених інфекційних захворювань. Найімовірнішим пусковим механізмом кровотечі на тлі інфекції є порушення проникності судин. Внаслідок цього з’являються самочинні кровотечі діапедезного типу.
Гемартрози великих суглобів виникають тим раніше, чим тяжчою є форма гемофілії. Із загальної кількості крововиливів гемартрози становлять 70-80%, гематоми – 10-20%, крововиливи до центральної нервової системи – менше 5%, гематурія – 14-20%, шлунково-кишкові крововиливи – близько 8%.
ГЕМАРТРОЗ
Гемартроз – найчастіший і найспецифічніший прояв гемофілії. Найчастіше перші гемартрози виникають у віці 1-8 років і є наслідком травми. Гострий гемартроз супроводжується больовим синдромом, зумовленим підвищенням внутрішньосудинного тиску. Суглоб збільшено в об’ємі, шкіра над ним гіперемована та гаряча на дотик. За великих крововиливів може виявлятися флуктуація. Якщо гемартроз виникає після травми, слід виключити додаткові пошкодження (внутрішньосуглобовий перелом, відрив виростка, защемлення тканин).
Перебіг гемофільної артропатії включає три фази:
– гострий крововилив до порожнини суглоба, виражений больовий синдром, обмеження рухів, на рентгенограмах кісткові зміни не виявляються;
– синовіт, патологічні зміни синовіальної оболонки, суглобового хряща;
– формування деформуючого остеоартрозу та контрактур.
За тяжкої форми гемофілії більшість гемартрозів не пов’язано з фізичними навантаженнями та травмами, а виникають самочинно. За гемофілії середньої тяжкості гемартрози виникають, зазвичай, після травми, але можуть розвиватися й спонтанно. За легкої форми гемофілії гемартрози, зазвичай, розвиваються на тлі серйозних забоїв і травм. Рецидивуючі гемартрози викликають хронічний синовіт і хронічні геморагічно-деструктивні остеоартрози.
На стадії синовіту синовіальна оболонка гіпертрофується і стає основним джерелом крововиливу до суглоба. В разі гострого синовіту гемартрози можуть виникати один за одним попри трасфузії дефіцитних чинників, що пов’язано із запальним процесом у синовіальній оболонці. При цьому у хворих часто (4-5 разів на місяць) виникає сильний біль, що має нетривалий характер. За хронічного синовіту біль у суглобі може бути відсутнім, оскільки зруйновано капсулу суглоба, але суглоб залишається постійно збільшеним в об’ємі.
Деформуючий остеоартроз формується у хворих на пізніх стадіях патологічного процесу, коли відбувається фіброзне переродження синовіальної оболонки. Ексудат у суглобі більше не накопичується. Загострення гемартрозів трапляються рідше, рухливість у суглобі прогресивно зменшується. У малорухливих колінних суглобах крововиливи у вигляді окремих, оточених фіброзною капсулою кіст, локалізуються у верхньому та бічних заворотах, у ділянці міжвиросткової ямки, а іноді і в товщі гіпертрофованої капсули. Кістозні зміни призводять до пошкодження кісткових стуктур та подальшого прогресування патологічного процесу.
На підставі клініко-рентгенологічних даних геморагічно-деструктивні остеоартрози поділяють на V стадій:
І. Рання стадія характеризується збільшенням об’єму суглоба, розширенням суглобової щілини за рахунок крововиливу. Рентгенологічно можуть виявлятися потовщення та ущільнення суглобової капсули, помірний остеопороз. Функцію суглоба за відсутності крововиливу у «холодний» період не порушено.
ІІ. Стадія характеризується помірним звуженням суглобової щілини без порушення конгруентності суглобових поверхонь. Наростають ознаки остеопорозу. З’являється субхондральний склероз. Відбувається подальше ущільнення периартикулярної тканини.
ІІІ. Стадія характеризується появою крайових узур, деструкцією хрящової тканини з утворенням кіст. Остеопороз більш виражений. Суглобову щілину звужено, місцями порушено конгруентність суглобових поверхонь. У колінному суглобі відбувається характерна зміна надколінника – квадратна форма його нижнього полюсу та збільшення передньозаднього розміру. Функцію суглоба помірно знижено, незначно обмежено рухи, є атрофія м’язів.
IV. Стадія, на якій суглоби різко деформовано, суглобові поверхні сплющено, епіфізи розширено за рахунок гіперостозів, діафізи зменшено, суглобову щілину різко звужено. Внутрішньосуглобові хрящі зруйновано. Виражена атрофія м’язів. Обсяг рухів значно обмежено. Відзначається внутрішньосуглобова крепітація під час руху. Функцію суглоба значно порушено.
V. Стадія характеризується цілковитою втратою функції суглоба. Суглобова щілина погано контурується на рентгенограмі, часто вона заросла сполучною тканиною. Виражено склероз субхондральних відділів кістки, приєднано значну узурпацію та кістоз епіфізів. Відбувається формування кісткового анкілозу.
Деформуючі остеоартрози чинять вплив на динаміку всього опорно-рухового апарату: відбуваються зміни постави хворого, викривлення хребта і таза, розвиваються гіпотрофії м’язів та остеопороз. Тривала неправильна постава стопи спричинює вальгусну деформацію колінного суглоба та утворення сталої контрактури у скоковому суглобі за типом «кінської стопи».
За використання милиць відзначаються крововиливи до суглобів верхніх кінцівок. Глибокі зміни кістково-суглобової системи обумовлюють настання інвалідності вже у дитячому віці.
Вторинний ревматоїдний синдром (синдром Баркагана-Єгорової) нашаровується на попередні гемартрози та властиві гемофілії деструктивні процеси у суглобах. Цей синдром супроводжується хронічним запальним процесом у дрібних суглобах кистей і стоп із наступною їх деформацією. Відзначається скутість, персистуючий біль у суглобах. У більшості хворих синдром проявляється у віці понад 14 років. Із віком тяжкість ураження суглобів прогресує, що призводить до обмеження рухової функції, формування контрактур та анкілозів.
Гематоми – крововиливи до м’яких тканин найчастіше локалізуються у ділянці м’язів, що несуть на собі найбільше статичне навантаження (здухвинно-поперековий, чотириголовий м’яз стегна, триголовий м’яз гомілки). Обширні гематоми можуть досягати значних розмірів, викликати анемію у хворого. Крім того, великі гематоми супроводжуються компресією оточуючих тканин. Гематоми, тиснучи на нервові стовбури або м’язи, викликають порушення чутливості, атрофію м’язів і контрактури.
Можливі патологічні переломи довгих трубчастих кісток внаслідок атрофічних і кістозних змін, які також супроводжуються простяжними гематомами, що розшаровуються. За крововиливів у ділянку здухвинного м’яза формується згинальна контрактура стегна. Простяжні крововиливи до м’яких тканин підщелепної ділянки, шиї, зіва та глотки створюють небезпеку стенозування верхніх дихальних шляхів та асфіксії.
За адекватного інтенсивного лікування гематоми цілком розсмоктуються у більшості хворих. В окремих випадках гематоми можуть трансформуватися у «гемофілічні псевдопухлини». У цих випадках проводиться хірургічне лікування.
Гематурія може виникати спонтанно або у зв’язку з травмами поперекової ділянки. Гематурія може супроводжуватися дизурічними проявами, нападами ниркової кольки, обумовленими утворенням згустків крові у сечовивідних шляхах. На підставі дослідження нирок у хворих на гемофілію можна знайти такі нефрологічні порушення, як нирковий капілярний некроз, гідронефроз, пієлонефрит. Макрогематурію у хворих на гемофілію зумовлено наявністю конкрементів у сечовому міхурі, пієлоектазією, гідронефрозом. Проте не завжди вдається знайти причину гематурії. Вона може бути іноді єдиним симптомом початкової стадії гідронефрозу, виникаючи внаслідок раптового та швидкого зниження внутрішньомискового тиску. Надто тяжко гематурія перебігає у хворих з інгібіторною формою гемофілії. Діагностика причин гематурії у хворих на гемофілію дозволяє визначити тактику подальшої терапії – як консервативної, так і оперативної.
Шлунково-кишкові кровотечі у хворих на гемофілію не є переважною формою кровоточивості. Профузні шлунково-кишкові кровотечі за гемофілії бувають спонтанними, можуть бути викликаними прийманням ацетилсаліцилової кислоти, інших нестероїдних протизапальних засобів. Крім того, джерелом кровотечі є латентні виразки шлунку та дванадцятипалої кишки, а також ерозійні гастрити, гемороїдальні вузли.
Крововиливи до брижи та сальника можуть імітувати гостре хірургічне захворювання органів черевної порожнини (гострий апендицит, кишкова непрохідність тощо).
Крововиливи до головного та спинного мозку та їх оболонок за гемофілії трапляються через травму. В окремих випадках причиною таких крововиливів може бути гіпертонічний криз або приймання препаратів, що значно порушують гемостатичну функцію тромбоцитів (ацетилсаліцилова кислота, бутадіон тощо).
Поява осередкової симптоматики у хворого на гемофілію, який зазнав травми голови, вимагає термінового призначення антигемофільних препаратів, подальшого лікування в умовах стаціонару під наглядом невропатолога. Будь-який хворий на гемофілію із симптоматикою, що свідчить про можливий крововилив до головного або спинного мозку, включаючи сонливість або незвичайний головний біль, потребує термінової госпіталізації.
У хворих на гемофілію можуть спостерігатися тривалі кровотечі в разі травм або операцій. Найнебезпечнішими є рвані рани. Кровотечі після пошкодження м’яких тканин часто виникають не одразу, а через деякий час (1-5 годин). Усі хірургічні втручання у хворих на гемофілію, включаючи діагностичні інвазійні процедури (пункційна біопсія), проводяться із застосуванням гемостатичної терапії препаратами – чинниками зсідання VIII або ІХ. Видалення до 3 зубів, окрім молярів, проводиться в амбулаторних умовах на тлі гемостатичної терапії. Множинне або технічно складне видалення зубів здійснюється у стаціонарних умовах. У хворих на гемофілію, ускладнену наявністю інгібіторів, видалення зубів проводиться в умовах стаціонару, можна видаляти щонайбільше один зуб. У зв’язку з підвищеним ризиком будь-якої місцевої анестезії рекомендовано застосування загальної анестезії.
Підозра на гемофілію та необхідність її діагностики виникає за будь-якої тривалої кровотечі, незалежно від її локалізації (з пуповини, кефалогематом у новонароджених, після видалення зубів та оперативних втручань у дорослих).
Діагноз гемофілії встановлюється на підставі відповідних клінічних проявів, генеалогічних даних, результатів дослідження показників гемостазу:
– подовження активованого часткового (парціального) тромбопластинового часу (АЧТЧ);
– зниження прокоагулянтної активності чинників зсідання VIII або ІХ нижче від 50%.
Організація надання допомоги хворим на гемофілію здійснюється лікарями-гематологами у тісній співпраці із педіатрами, терапевтами, сімейними лікарями, травматологами, хірургами, стоматологами та лікарями інших спеціальностей.
Станіслав ВИДИБОРЕЦЬ
Світлана ГАЙДУКОВА
Галина МОРОЗ
Олександр СЕРГІЄНКО
Національна медична академія
післядипломної освіти ім.П.Л.Шупика
Бажаєте знати більше – прочитайте:
1. Баркаган З.С., Момот А.П. Диагностика и контролируемая терапия нарушений гемостаза. М.: Ньюдиамед, 2001.-С.70-117.
2. Влияние современной заместительной терапии на качество жизни детей с гемофилией А / И.И. Спичак, Е.В. Жуковская, Е.В. Барашова и соавт. // Гематол. и трансфузиол.-2009.-Т.54,№2.-С.27-31.
3. Гемофилия у детей: эффективность физиотерапевтических факторов в комплексном лечении / К.И. Григорьев, Г.И. Сосков, А.А. Григорьев и соавт. // Мед. помощь.-2008.-№6.-С.3-6.
4. Румянцев А.Г., Чернов В.М., Никаноров А.Ю. Современные достижения и проблемы в лечении гемофилии // Вопр. гематол. онкол. и иммунопатол. в педиатр.-2003.-Т.2, №2.-С.7-14.





